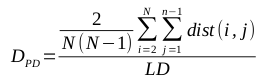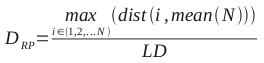A brief primer on HMMs
I think that HMMs are best described by an example. We will look at this toy model:

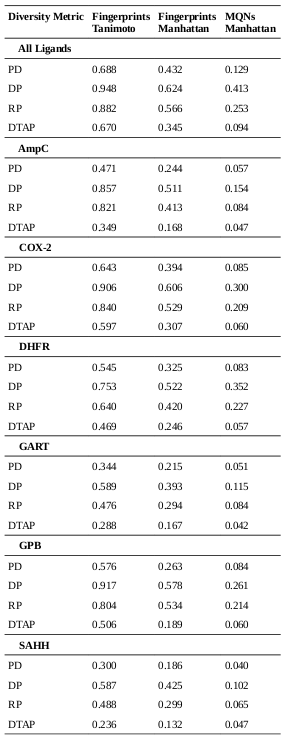
The numbers represent the probabilities of transitioning between the various states, or of emitting certain symbols. For example, state S1 has a 90% chance of transitioning back to itself; each time it is visited, there is a 50% chance that it emits a ‘1’, and a 50% chance that it emits a ‘2’.
Clearly, this model can be used to produce strings of 1s and 2s that fit its parameters. But we can use it to solve a much more interesting question: given a string of 1s and 2s, which sequence of states most likely generated the string? This is why it’s described as a hidden Markov model; the states that were responsible for emitting the various symbols are unknown, and we would like to establish which sequence of states is most likely to have produced the sequence of symbols.
Let’s look at what might have generated the string 222. We can look at every possible combination of 3 states to establish which was most likely responsible for that string.
- S1 – S1 – S1: 0.5 (initial probability of being in state 1) * 0.5 (probability of S1 emitting a 2) * 0.9 (probability of S1 transitioning to S1) * 0.5 (probability of S1 emitting a 2) * 0.9 (probability of S1 transitioning to S1) * 0.5 (probability of S1 emitting a 2) = 0.050625
- S1 – S1 – S2: 0.5 * 0.5 * 0.9 * 0.5 * 0.1 * 0.75 = 0.0084375 (less likely than the previous sequence)
- S1 – S2 – S1: 0.5 * 0.5 * 0.1 * 0.75 * 0.8 * 0.5 = 0.0075
- S1 – S2 – S2: 0.5 * 0.5 * 0.1 * 0.75 * 0.2 * 0.75 = 0.0028125
- S2 – S1 – S1: 0.5 * 0.75 * 0.8 * 0.5 * 0.9 * 0.5 = 0.0675
- S2 – S1 – S2: 0.5 * 0.75 * 0.8 * 0.5 * 0.1 * 0.75 = 0.01125
- S2 – S2 – S1: 0.5 * 0.75 * 0.2 * 0.75 * 0.8 * 0.5 = 0.0225
- S2 – S2 – S2: 0.5 * 0.75 * 0.2 * 0.75 * 0.2 * 0.75 = 0.0084375
The string 222 was most likely generated by the sequence S2 – S1 – S1. That may be a little surprising, since S2 is much more likely than S1 to emit 2s, but the transition probabilities show that the model is generally much more likely to be in state S1.
When the string of observations to explain is very long, this enumeration of every possible sequence of states becomes infeasible. We had to enumerate 2^3 possible sequences for a string of length 3; if the string were of length 400, we would need to enumerate 2^400 (about 2.6 * 10^120) sequences. Instead, we can employ a dynamic programming approach to make the problem tractable; the module that I wrote includes an implementation of the Viterbi algorithm for this purpose.
Why is this interesting?
A good example of the utility of HMMs is the annotation of genes in a genome, which is a very difficult problem in eukaryotic organisms. A gene typically consists of a promoter region, numerous exons and introns with their associated splice sites, and a poly-A region, among others. A model can be created to describe each of these regions. The most likely sequence of states that explains a genome can then be calculated. Each nucleotide in the genome would be annotated with a probable state, indicating whether it is likely to be part of an intron, exon, splice site, intergenic region, etc.
There are a wide variety of real-world applications for HMMs, such as in signal processing, or in identifying secondary structure elements in proteins, parts of speech in sentences, or components of musical scores.
The hmm Python module
With my Python module, the above model can be created with the following:
import hmm
s1 = hmm.state(
'S1', # name of the state
0.5, # probability of being the initial state
{ '1': 0.5, # probability of emitting a '1' at each visit
'2': 0.5 }, # probability of emitting a '2' at each visit
{ 'S1': 0.9, # probability of transitioning to itself
'S2': 0.1 }) # probability of transitioning to state 'S2'
s2 = hmm.state('S2', 0.5,
{ '1': 0.25, '2': 0.75 },
{ 'S1': 0.8, 'S2': 0.2 })
model = hmm.hmm(['1', '2'], # all symbols that can be emitted
[s1, s2]) # all of the states in this HMM
All of the possible paths explaining 222 can be generated with:
model.enumerate('222')
Which would print the following:
('S2', 'S2', 'S2'): -2.073786
('S2', 'S2', 'S1'): -1.647817
('S2', 'S1', 'S2'): -1.948847
('S2', 'S1', 'S1'): -1.170696
('S1', 'S2', 'S2'): -2.550907
('S1', 'S2', 'S1'): -2.124939
('S1', 'S1', 'S2'): -2.073786
('S1', 'S1', 'S1'): -1.295635
BEST: ('S2', 'S1', 'S1'): -1.170696
Note that the probabilities are log (base 10) transformed, as they will be very tiny numbers for long strings.
As stated above, don’t do that with long sequences! It becomes intractable very quickly. Instead, use the dynamic programming approach as follows:
path, prob = model.viterbi_path('222')
print path
print prob
Which yields:
['S2', 'S1', 'S1'] -1.17069622717]]>